E&E PhD Exit Webinar: Systematic bias in phylogenetic inference: Implications, Assessment, and Reduction
E&E PhD Exit Webinar: Systematic bias in phylogenetic inference: Implications, Assessment, and Reduction
Phylogenetic inference is the process of reconstructing relationships between species from genomic sequence data. The reliability of phylogenetic analysis relies on the quality of the data and the fitness of the substitution models.
Speakers
Event series

Content navigation
Description
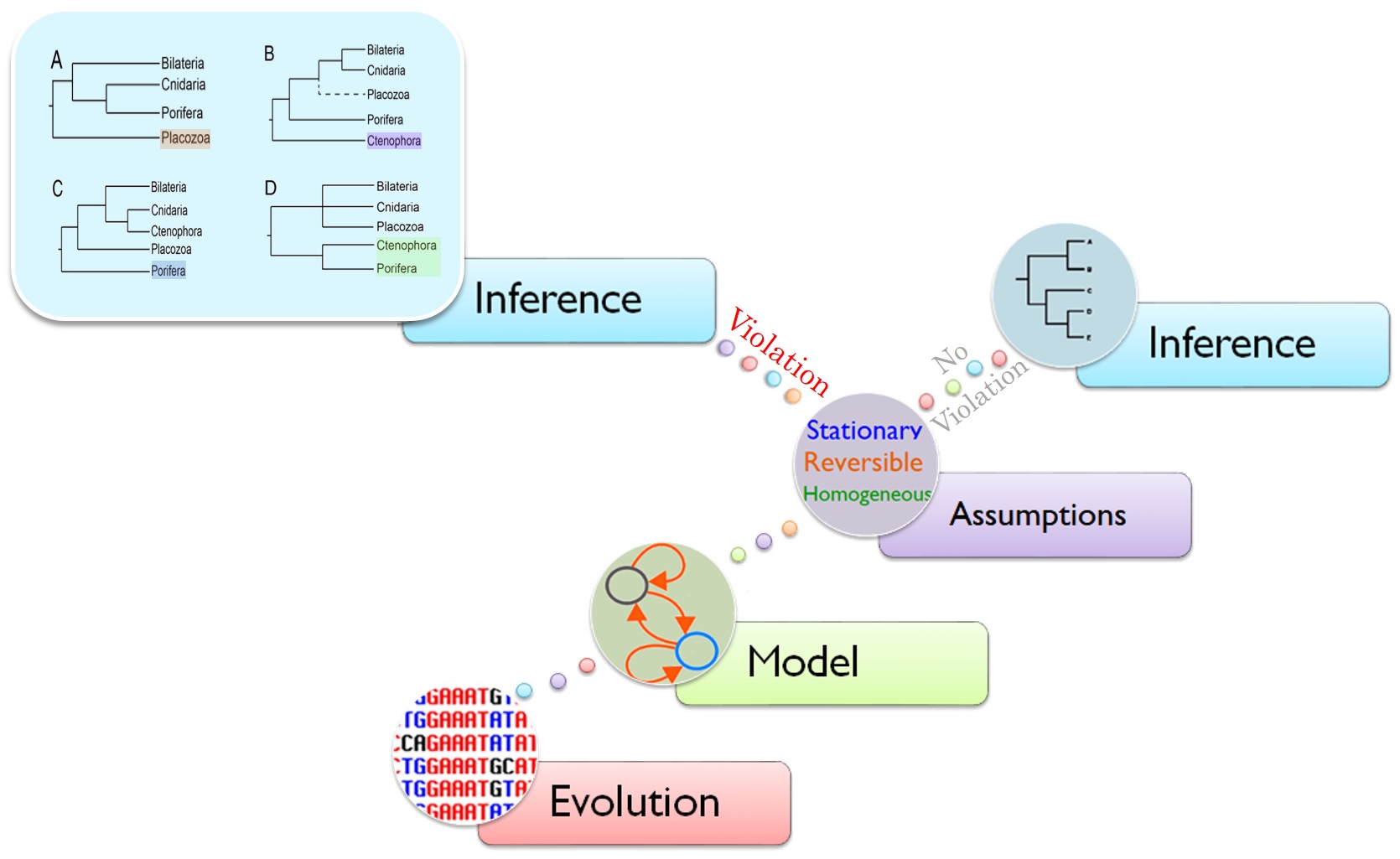
Phylogenetic inference is the process of reconstructing relationships between species from genomic sequence data. The reliability of phylogenetic analysis relies on the quality of the data and the fitness of the substitution models. In phylogenetic inference, we commonly use substitution models which assume that sequence evolution is stationary, reversible, and homogeneous (SRH). Many empirical and simulation studies have shown that assuming SRH conditions can lead to significant errors in phylogenetic inference when the data violates these assumptions. Yet, the extent of SRH violations and their effects on phylogenetic inference of tree topologies are not well understood. Moreover, using time-reversible Markov models can produce unrooted phylogenetic trees only.
In my thesis, I introduce and apply new tests to assess the scale and impact of SRH violation on empirical and simulated datasets. In addition, I investigate the utility of non-reversible models to root empirical phylogenetic trees and introduce new test statistics, which provide information on the statistical support for any given root position.
Location
Please click the link below to join the webinar:
https://anu.zoom.us/j/81457198125?pwd=OUYybjEycmNIVG5NaUpvRkNROXRMQT09
Passcode: 416746
Canberra time: please check your local time & date if you are watching from elsewhere.